









: Symptoms and Treatment")
Acid sphingomyelinase deficiency, or ASMD, is a rare inherited disorder that affects how the body breaks down fats. It leads to a harmful buildup inside cells, especially in the liver, spleen, lungs, and sometimes the brain. Once known as Niemann-Pick disease types A and B, ASMD impacts roughly 1 in 250,000 births in the United States, with an estimated fewer than 200 people living with it nationwide. For families in the U.S., the condition can feel overwhelming; symptoms often start in childhood and worsen over time if untreated.
The encouraging news? Since the FDA approved the first targeted treatment in 2022, early diagnosis and care can dramatically improve quality of life and even extend survival. This guide explains ASMD in clear, straightforward terms so you can spot the signs, understand the science, and explore real options. Whether you’re a parent worried about a child’s unexplained symptoms or an adult living with chronic issues, knowledge is the first step toward better health.
What is ASMD?
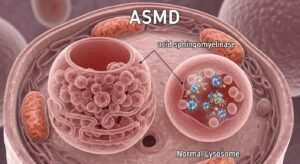
ASMD is a lysosomal storage disorder caused by a shortage of the enzyme acid sphingomyelinase (ASM). Without enough ASM, a fatty substance called sphingomyelin builds up in cells and damages organs over time. It’s progressive, meaning symptoms usually get worse without intervention, but it’s not contagious, and it affects people of all backgrounds.
In the U.S., it’s considered ultra-rare, yet awareness is growing because new therapies are changing outcomes. ASMD is now viewed as a spectrum rather than strictly separate diseases, ranging from severe infantile forms to milder adult-onset cases.
Symptoms
Symptoms of ASMD vary widely depending on the type and age of onset, but they often involve multiple organs. Early recognition can make a big difference.
Visceral (Organ) Symptoms
The most common signs include an enlarged liver and spleen (hepatosplenomegaly), which can cause a swollen belly, easy bruising, and low platelet counts, leading to bleeding risks. Many people also develop interstitial lung disease, resulting in shortness of breath, frequent infections, or reduced oxygen levels. Growth delays are frequent in children, with slower height and weight gain despite a good appetite.
Blood and Digestive Issues
Thrombocytopenia (low platelets) and anemia often appear, increasing fatigue and bruising. Digestive problems like diarrhea, poor nutrient absorption, or elevated cholesterol and liver enzymes are also typical and can affect daily energy levels.
Neurological Symptoms in Severe Cases
In more serious forms, brain involvement leads to developmental delays, loss of motor skills, seizures, or cherry-red spots in the eyes visible on exam. These are less common in milder types but signal the need for urgent specialist care.
Causes
ASMD stems from specific genetic changes that disrupt normal cell cleanup.
Genetic Mutations in the SMPD1 Gene
Mutations in the SMPD1 gene reduce or eliminate ASM enzyme activity, often to less than 10% of normal levels. This is an autosomal recessive condition, meaning a child must inherit one faulty copy from each parent.
How Sphingomyelin Buildup Happens
Without working ASM, sphingomyelin and other lipids accumulate in lysosomes (the cell’s recycling centers). Over the years, this causes inflammation, scarring in the lungs, and organ enlargement that strains the body.
Inheritance and Risk Factors
Carriers (with one mutated gene) have no symptoms but pass the risk to children. Type A is more common in people of Ashkenazi Jewish descent, but all ethnic groups can be affected. Family history or consanguinity raises the odds.
Treatment Options
There is no cure for ASMD yet, but targeted therapy and supportive care have transformed management since 2022.
Enzyme Replacement Therapy (ERT) with Xenpozyme
Olipudase alfa (Xenpozyme), approved by the FDA in August 2022, is the first and only treatment for non-central nervous system symptoms in both adults and children. It replaces the missing ASM enzyme, reducing spleen and liver size by up to 78% and 60%, respectively, in long-term studies (up to 8 years). It also improves lung function (diffusing capacity often rises over 50%), growth in kids, and overall quality of life with mostly mild side effects.
Supportive and Multidisciplinary Care
Regular monitoring by a team, including hematologists, pulmonologists, and dietitians, helps manage complications. This may include blood transfusions, respiratory support, pain relief, physical therapy, and nutritional plans to support growth.
Emerging Research and Clinical Trials
Gene therapy and other disease-modifying approaches are in development. Clinical trials continue to explore better dosing and combination treatments, offering hope for even more options in the coming years.
Types of ASMD
ASMD exists on a spectrum, but doctors often describe three main forms based on age, severity, and neurological involvement.
Type A: Infantile Neurovisceral (Most Severe)
This form appears in early infancy with rapid brain involvement, severe organ enlargement, and developmental regression. Without treatment for non-brain symptoms, most children do not survive past age 3.
Type B: Chronic Visceral (Milder, Non-Neurological)
Symptoms start in childhood or later and mainly affect the liver, spleen, lungs, and blood. Many people live into adulthood with proper care, though lung disease and growth issues can limit daily activities.
Type A/B: Intermediate (Chronic Neurovisceral)
This overlaps the two extremes, with some neurological symptoms alongside visceral problems. Progression is slower than Type A but faster than pure Type B, requiring close monitoring of both body and brain function.
When to Seek Medical Care for ASMD
Contact a doctor right away if a child shows persistent belly swelling, unexplained bruising, breathing difficulties, or failure to meet developmental milestones. Adults with ongoing fatigue, shortness of breath, or unexplained organ enlargement should also seek evaluation, especially with a family history of rare diseases. Early intervention with specialists can prevent complications and qualify patients for approved therapies.
Diagnosis
Diagnosis combines clinical clues with lab tests. Doctors start with blood work showing low ASM enzyme activity (usually via dried blood spot or white blood cells). Confirmatory genetic testing for SMPD1 mutations is standard and available in the U.S. through specialized labs. Imaging (ultrasound or CT) checks organ size, while lung function tests and eye exams help assess severity.
Some states include ASMD in newborn screening, which can lead to diagnosis before symptoms appear. The average diagnostic delay is around 5 years, so advocating for parallel testing with similar conditions like Gaucher disease speeds things up.
How to Prevent Future Danger of ASMD
While you can’t prevent the genetic mutation itself, families can reduce risks through planning.
Genetic Counseling and Carrier Screening
Couples with a family history or from higher-risk groups should get carrier testing before pregnancy. Genetic counselors explain odds and options like prenatal testing or preimplantation genetic diagnosis (PGD) during IVF.
Family Planning and Newborn Screening Awareness
Knowing your status empowers informed decisions. In the U.S., check your state’s newborn screening panel; early detection allows prompt treatment with Xenpozyme to limit damage.
When to See a Doctor
See a healthcare provider if you notice any combination of enlarged abdomen, frequent respiratory infections, easy bleeding, growth concerns in children, or unexplained fatigue that doesn’t improve.
For those already diagnosed, schedule regular follow-ups every 6-12 months with a metabolic specialist to track progress and adjust care. Don’t wait for symptoms to worsen; U.S. rare disease centers offer coordinated expertise that can change the trajectory.
FAQs
What Is ASMD and How Does It Affect Patients?
ASMD is a genetic enzyme deficiency causing fat buildup in organs. It leads to enlarged liver/spleen, lung problems, growth delays, and, in severe cases, neurological decline, but targeted therapy now helps many patients live fuller lives.
Why Is Early Diagnosis of ASMD Crucial?
Early diagnosis prevents irreversible organ damage and allows starting Xenpozyme before complications become severe. Studies show better lung function, growth, and survival when treatment begins in childhood.
When Should You Seek Treatment for ASMD?
Seek treatment as soon as the diagnosis is confirmed, especially for non-brain symptoms. Xenpozyme is approved for all ages and works best when started early to slow progression.
Who Is Most Affected by ASMD?
It affects males and females equally across ethnicities, though Type A is more frequent in Ashkenazi Jewish populations. Most U.S. cases are diagnosed in childhood, with Type B being the most common overall.
How Can ASMD Impact Daily Life?
It can cause chronic fatigue, breathing limitations, frequent medical visits, and school or work challenges. With modern care, many patients manage symptoms well and maintain independence, though lifelong monitoring is usually needed.
Conclusion
Living with or caring for someone with ASMD used to mean limited options, but today’s advances, like FDA-approved Xenpozyme and growing specialist networks, bring real hope. By understanding the symptoms, causes, and treatments, families across the United States can take control and advocate effectively.
If you suspect ASMD in yourself or a loved one, talk to your doctor or contact a rare disease center right away. Share this article to raise awareness, and remember: early action truly saves lives. Stay informed, stay connected, and never hesitate to seek the support you deserve.



